Aqsa Sarwara, Rabia Nawaza’b*, Saba Batoola, Muhammad Shahidb, Areena Aftaba, Kainat Ahmeda, Aneesa Naseera, Anum Ajmalc, Uqba Mehmooda
aDepartment of biological sciences, Superior University, Raiwind road, Lahore.
bCenter of excellence in molecular biology, university of the Punjab, Lahore.
cDepartment of Zoology, University of the Punjab, Lahore.
Received: 19-Jan-2022 / Revised and Accepted: 2-March-2022 / Published On-Line: 01-May-2022
Abstract:
HCV is a life-threatening disease that causes several liver disorders such as liver fibrosis, cirrhosis, and hepatocellular carcinoma (HCC)-second leading type of cancer. Although directing acting antivirals (DAAs) are showing some promising results, despite the liver complications caused by HCV are likely to persist in near future. HCV induced HCC is a slowly progressing multiple step process. This carcinogenesis is acquired over the years via production of a cirrhotic tissue microenvironment. Some cellular proteins and eukaryotic translation initiators were seen to increase translation of HCV polyprotein precursor and hence intracellular replication of viral gene. Which in turn upsets the regulation of several cytokines leading to cytokine storm in the liver and peripheral blood mononuclear cells (PBMCs). This plays a role in liver disease progression, thus signifying role of PBMCs in HCC induction over time.
Keywords: HCV, HCC, PBMC, disease progression, liver
Introduction:
Hepatitis C virus (HCV) is the major factor causing severe hepatitis and liver disease [1]. HCV affects over 170 million people worldwide and over 16 million in Pakistan. HCV infection spreads mostly through blood-to-blood contact [2]. HCV serum was isolated from the person with
none-A and none-B hepatitis [3]. Choo et al in 1989 described the family of HCV; he explained that it belongs to the family of Flabiviridae [4]. In the cytoplasm of hepatocytes, HCV can cause infection and regulate but it is not cytopathic rapid production and continuous cell-cell separating may cause infection along with the absence of T cells immune response to HCV, the rate of HCV replication is between 10 raised to power 10 to 10 raised to power, 12 virions per day, and their estimated half-life is 2 to 3 hours [5]. They have 50 sub-types from the six known genotypes [6].
During the beginning of HCV, it was considered that a patient who progresses jaundice symptoms is more likely to have less HCV infection as opposed to those who are without jaundice or unhealthy [7]. Development of cirrhosis from the HCV is often clinically silent and some patients are known at the time when the cirrhosis convert in the ending stage Hepatocellular carcinoma (HCC), the characteristics of decompensated cirrhosis involve production of ascites, upper GI (gastro intestinal) bleeding secondary to varicose veins (large tortuous veins found on legs), hypertensive(allergy) gastropathy hepatorenal syndrome and hepatic encephalopathy [8]. It’s uncertain if direct-acting antiviral medications (DAAs) totally eliminate HCV infection or any virus may remain after a durable virologic response has been achieved [9].
In infected individuals, the hepatitis C virus is good enough to stay in the host and cause permanent liver damage [10]. Infection CLTs were found in both HCV-related and unconnected chronic infections, indicating that this characteristic is not unique to HCV and is found in a wide range of lasting viral infections [11], [12], [13].
HCV has a lot of genetic variation, and it’s divided into a few primary genotypes that vary by even more than 30% [14]. The absence of an efficient immunological response in HCV infection is due to HCV’s ability to quickly change its envelope proteins in order to evade the nullifying antibody response in individuals [15], [16], [17].
HCV was first described in January 2004 by Castillo et [18]. The livers of a hundred patients were found to be abnormal and could be tested. Alanine: Amino transferase or gamma-glutamyl trans peptidase was generated as a result of the test. In a reverse transcription polymerase chain reaction, it was discovered that half of them contained hcv-RNA in their liver.
As a result, Stapleton and colleagues have described a number of readings on sero-negative HCV infection in patients with cryptogenic liver disease, which can lead to aberrant liver test results [19]. In PBMC cells, HCV plays a significant role [20]. Antigenic HCV-RNA, which can be synthesized by Reverse Transcription Polymerase Chain Reaction (rt-PCR), was found in 61 percent of patients’ PBMCs. These can detect the presence of HCV in PBMC cells [21].
HCV induced Disease Progression:
Hepatitis C virus (HCV) is one of the most common causes of hepatocellular cancer (HCC). HCV’s connection with its human host is complicated, and multifaceted, due in part to the fact that it is an RNA virus that cannot incorporate into the host’s DNA. Activation of numerous host routes, such as liver fibro-genic routes, cellular and survival routes, and contact to immunological and metabolic processes, are all direct and indirect processes of HCV-induced HCC. As indicated by genomic research proving polymorphisms in immunological, metabolic, & growth regulatory pathways linked to an enhanced danger of HCC, host variables are important in HCV-induced HCC. The mortality and frequency of HCV-related liver diseases, such as HCC, is expected to continue in the coming years despite the accessibility of very efficient direct-acting antivirals. In order to identify HCV patients at increased danger of growing HCC have also been described; although, this would need to be confirmed extensively, particularly in those who have had a prolonged virologic therapy. Clinical systems are beginning to incorporate molecular indicators that encourage greater improvement of HCC danger, enabling for objective risk assessment and personalized medication and monitoring for HCV patients. Enrichment of HCC chemoprevention clinical trials might also benefit from molecular indicator-based classification, resulting in lesser sample as well as lower charges.
Hepatocellular carcinoma (HCC) is a major scientific concern, since it is the world’s 2nd largest reason of cancer fatalities [22] and it is also one of the few forms of cancer-related death which is expanding in the United States [23]. A leading cause of HCC in developed countries is hepatitis B virus (HBV) infection, chronic infection with hepatitis C virus (HCV), in comparison to developing countries in the Asia-Pacific area and Sub-Saharan Africa [24]. In the United States, for HCC patients, it is the primary evidence for liver transfer [25].
It is predicted that almost 3% of population has been infected with HCV according to the World Health Organization (WHO). About more than 170 million individuals were actively carrying the virus as chronic carriers [26]. In the United states, HCV-infected the individuals that were born amid 1945 and 1965 were named “baby boomers” and they were more than 75% [27].
Hepatitis C has evolved into a worldwide health threat over time. A variety of physical, genetic, and epigenetic changes can be included in its harmful effects on the human liver. Fatty liver also known as hepatic steatosis is an inflammatory condition with several causes, one of which being steatohepatitis. HCV stimulates a multitude of biological processes, including the upregulation of cytokines. The regulation of a few important cytokines during HCV infection in order to gain a better knowledge of their function is discussed here. These cytokines, IL-1β, IL-6, TNF-α, and IFN-ϒ, are known the inflammatory indicators of the body. These and other indicators aid hepatocytes in their fight against viral infection. However, their link has recently been discovered in liver degeneration on the path to non-alcoholic steatohepatitis (NASH). As a result, disruptions in their equilibrium have been recorded often during HCV infection. A few of studies has confirmed up-regulation of cytokines. Although hepatocytes activate these cell markers as a conventional defensive strategy, recent research has shown the contradictory nature of this defense line. However, direct molecular or epigenetic study is required to investigate the molecular pathways that lead to steatosis, cirrhosis, or even HCC in the liver (Hepatocellular Carcinoma) [28]
Role of interleukin in liver disease progression:
Viral hepatitis, alcohol usage, and non-alcoholic steatohepatitis are the three primary causes of liver inflammation and chronic damage. These can progress to liver fibrosis, cirrhosis, and hepatocellular cancer, which may need a liver transplant. The interleukin (IL)-20 subfamily of the interleukin (IL)-10 family of cytokines aids the liver’s response to injury and illness, as well as the management of tissue homeostasis and the development of immune responses in this organ. The IL-22 cytokine is the most well-considered part of the family in the inflammatory balance of the liver, and it may have a protective function in fibrosis advancement while also increasing liver tissue sensitivity to hepatocellular carcinoma formation. Because certain members of the family share IL receptor subunits and signal through common intracellular pathways, other members of the family may likewise perform this double role. In the case of liver illness, researchers are beginning to examine targeting members of the IL-20 subfamily. The involvement of miRNA in the transcriptional regulation of IL-22 and IL-24, which was recently discovered, opens the door to potential new techniques for reducing organ harm and modulating the local immune response. In non-alcoholic steatohepatitis, the IL-20RA cytokine receptor has also been categorised as being under miRNA control. Furthermore, researchers have recommended that anti-inflammatory medications be combined with IL-22 as a hepatoprotective IL for the treatment of alcoholic liver disease (ALD), and clinical studies of ILs for the treatment of severe alcoholic-derived liver degeneration are now underway [29].
A common kind of liver damage known as alcoholic liver syndrome that can progress to fibrosis and cirrhosis worldwide. The National Institute on Alcohol Abuse published the current absorption study. It was discovered that liver cirrhosis was the 12th major cause of mortality in the United States in 2007, with a total of 29,925 fatalities. And 48 percent of these deaths were connected to alcohol. IL-22, on the other hand, has been found to protect mice against liver damage [30].
There is no longer any model of small animals obtained with prolonged viral inflammation of the liver that is linked to an increase in LL-22 in the liver, as enlightened earlier. IL-22 appearance in lymphoid cells and throughout the primary times after birth in transgenic mice [31]. The subfamily of IL-22 cytokine has a key role in inflammatory pathological conditions in the liver. In a mouse model of the IL-22 cytokine, the number of family members involved in liver homeostasis was carefully studied.
IL-22 also inhibits hepatitis stellated cell death and reduces liver fibrosis. IL-26 has antibacterial and antioxidant properties [32], [33], and it has been linked to hepatitis C (HCV) and liver fibrosis, as seen by eradicated branch hepatitis [34].
Elevated levels of Interleukin-22 increase the effects of liver tissue on the establishment of HCC. Physicians must evaluate the possibility of acquiring liver cancer if the findings are based on animal models. In the future, while considering treatment options for hepatitis patients. HCC cell metastasis has been demonstrated to be prevented by interleukin-29 [35].
In liver cell individuals with uncomplicated tumors, the production of interleukin 6 (IL-6) is elevated [36]. The amount of IL produced in hepatocytes and its concentration with in blood are linked to the severity of liver inflammation and fibrosis. Acute severe liver illness and tumor progression is induced by many pro-inflammatory cytokines (IL-1a,IL-1B, tumor necrosis, and IL-6) [37].
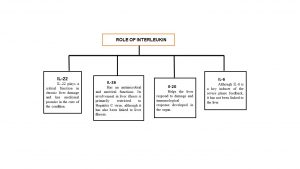
Figure 1: Role of interleukin-22, 26, 20 and interleukin-6 in liver disease progression
Role of miRNA in liver disease prog ression
In 1993, Ambros and colleagues discovered the miRNA [38]. In translation process miRNA binds to the RNAs [39]. Small RNAs are 18-24 nucleotides in length and also noncoding for chronic disease. In content of liver disease miR-34a, which is among the ideal miRNA, in early stages of liver fibrosis, analyzing miRNA is helpful to detect earlier and elevated levels in patients with fibrosis [40], [41].
In every organ miRNA have a specific expression and physiological situation, the expression of miRNA was described in content of liver disease [42], [43], [44]. In hepatic C, the patients with liver inflammation, such as alcoholic liver disease miR-155, has been described to up regulation [45].
In an experiment model of fibrosis in liver illness, miR-155 was found to be implicated in the advancement of liver inflammation. MiR-155 is still a mystery; however, it appears to serve a hepatic protective role in a non-alcoholic steatohepatitis model. The function of miRNA-155 in a variety of liver illnesses has gotten a lot of interest. In mice models of nonalcoholic steatohepatitis (NASH) and alcoholic steatohepatitis, introduction of miR-155 appearance has been seen [46], [47]. After consuming alcohol, the levels of miR-155 in Kupffer cells rose, and Tumor necrosis factor (TNF) has been recognized as an objective of miR-155 that encourages liver damage [47].
The most common miRNA in adult liver is miR-122. MiR-122 has been identified as an indicator for liver damage, and what’s even more intriguing is that miR-122 is a circulating blood protein [48], [49], [50]. MiR-122 has been found in the blood of people with Hepatitis B and C [51], [41], [52]. Include information about MiR-122 as a mechanism in liver damage, as well as the involvement of the HCC virus in hepatitis.
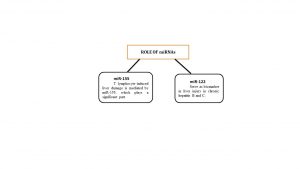
Figure 2: Role of miRNAs in liver disease progression
Direct-Acting Antivirals (DAAs) & Liver Fibrosis:
In chronic HCV, the incidence of HCC is linked to the severity of fibrosis. The yearly prevalence rate in cirrhotic patients is quite significant (1-7 percent annually), but HCC seldom occurs in livers that have less fibrosis. The incidence of HCV-related HCC can be lowered by the development of extremely effective direct-acting antivirals (DAAs), establishing a response called SVR (sustained virologic response) that doesn’t reduce the threat of HCC, particularly in individuals with symptomatic liver fibrosis [53].
HCV Replication in PBMCs:
Even if the complete process of how replication occurs in HCV is not known yet, it is thought that replication of HCV includes the formation of a negative strand RNA molecule that acts as a template for the formation of genomic HCV-RNA [54]. Therefore, presence of the negative strand of HCV-RNA proofs that viral replication occurs. The main site for virus replication is liver but some other sites have also been witnessed like peripheral blood mononuclear cells (PBMC) [55], [56], [57], [58]. Concerning about the infection of PBMC, HCV may spread in lymphoid cell cultures and the virus produced is infectious, according to research [59], [60]. Furthermore, it has been suggested that PBMCs may be the reason of persistent HCV infection following liver transplantation [61], [62]. HCV infections were recently discovered, which are considered as the occurrence of HCV-RNA in the liver in the absence of anti-HCV and blood HCV-RNA. In addition, up to 70% of these individuals carried HCV-RNA in their blood. PBMCs are the cells that make up a person’s blood [18]. Because these individuals have no detectable viral RNA in their blood, whether HCV replicates in PBMC is a key question in the transmission of occult HCV infection. Thus, using a strand specific reverse transcriptase-polymerase chain reaction (RT-PCR) and in situ hybridization, it was studied about HCV replication in PBMC of patients with occult HCV infection by detecting HCV-RNA positive and negative strands [63].
According to a study in China, HCV may infect and reproduce in PBMCs, which also contain the HCVNS5 protein. It was indicated that minus-strand HCV RNA in PBMCs might be one of the variables impacting IFN response as patients with minus strand HCV RNA in PBMCs had a substantially poorer 6-month sustained response to IFN treatment [64]. Despite the fact that hepatocytes are the primary location for the replication of HCV (hepatitis C virus), PBMC (peripheral blood mononuclear cells) have also been offered as a viable alternative [65] Another journal in Argentina concluded that HCV replication occurs in PBMCs [66]
In PBMCs from HCV-infected individuals, downregulation of STAT1 and IRF-1 expression and upregulation of caspase-3 expression may contribute to abnormalities in cytokine output and increased PBMC cell death observed in earlier research [67]
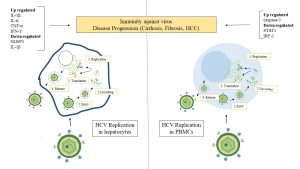
Figure 3: A schematic representation of PBMCs’ role in direct (via HCV replication) and indirect (via Cytokine, miRNA, caspase cascade activation) liver disease progression. The upregulation of IL-1β, IL-6, TNF-α, IFN-ϒ and downregulation of NLRP3, IL-1β during HCV replication in hepatocytes and upregulation of caspase-3 and downregulation of STAT 1, IRF-1 during HCV replication in PBMCs causes disease progression and plays a role in immunity against virus.
Though, it is not sure if apoptosis of virus-infected cells happens or not. Lin Deng et al reported in his study, that HCV infection caused cell death in Huh7.5 cells using chimeric J6/JFH1 strain of HCV genotype 2a. Cleavage of DNA repair enzyme (ADP-ribose) polymerase and activation of caspase 3, were all linked to cell death. These conclusions confirmed that HCV-induced cell death is apoptosis [68].
Interferons are present or detected in the liver as well as mononuclear cells of HCV patients’ peripheral blood [69]. This comprises three subgroups: il-29 (ifm-1), il-28aa (ifn-2), and il-28b (ifn-3), all of which cause hepatocytes to be stimulated by viruses or specific toll-like receptor ligands [70]. In HCV directed by HCV-specific cytotoxic t-lymphocytes response connected to viral clearance in chimps and humans during severe HCV infection Cd8 + t-cells play an important part [71].
Pathogen-induced immune responses can have a significant impact on all immune activation and resistance development [72]. The production of regulatory proteins by CD8+T cells, which results in fatigue, and mortality, activation of Foxp3+ regulatory T cells, as well as inhibition by interleukin-10 (IL-10) & other suppressive molecules, seem to be involved in this occurrence [73].
Conclusion
Hepatitis C virus (HCV) is one of the most prevalent causes of hepatocellular carcinoma (HCC), which is a major scientific concern since it is the second leading cause of cancer-related deaths in the world. HCV-related liver disorders, such as HCC, are likely to continue to increase in mortality and frequency. Hepatic steatosis, often known as fatty liver, is an inflammatory disorder caused by a variety of factors. HCV activates a wide range of cellular activities, including cytokine upregulation. Inflammatory markers such as IL-1, IL-6, TNF-, and IFN- are well-known in this regard.
MicroRNAs govern the activation of numerous genes and, as a result, affect a number of distinct functions, such as the type I interferon-mediated immunological response to infectious diseases (IFN). Role of different Interleukins and miRNA in disease progression is a well-known phenomenon. The interleukin (IL)-20 subfamily of the interleukin (IL)-10 family of cytokines aids the liver’s response to injury and illness. The IL-22 cytokine has a protective function in fibrosis advancement while it also increases liver tissue sensitivity for hepatocellular carcinoma formation. IL-22 also reduces liver fibrosis. The most common miRNA in adult liver is miR-122 and it has been identified as an indicator for liver damage. MiR-155 appears to serve a hepatic protective role in a non-alcoholic steatohepatitis model. Given this, HCV replication occurring in PBMCs, causing downregulation of STAT1 and IRF-1 expression and upregulation of caspase-3 expression may contribute to increased cell death in both PBMCs and hepatocytes, resulting in disease progression in liver. This also infers that some individuals that don’t have HCV-RNA in their serum/plasma on testing, might be infectious because HCV hides and multiplies in the PBMC of individuals with occult HCV infection.
Author’s Contribution:
R.N., Conceived the idea; R.N., Designed the simulated work. A.S., S.B., A.A.,A.A., K.A., and A.N., did the acquisition of data. A.S., S.B., Executed simulated work, review of literature and interpretation of data and wrote the basic draft; R.N., A.S., Did the language and grammatical edits. R.N., M.S., and U.M., did Critical revision. All authors have read and approved the manuscript.
Funding: The publication of this article was funded by no one.
Conflicts of Interest: The authors declare no conflict of interest.
REFERENCES
[1] Y.-C. Kwon, R. B. Ray, and R. Ray, “Hepatitis C virus infection: establishment of chronicity and liver disease progression,” EXCLI journal, vol. 13, p. 977, 2014.
[2] A. Khan, A. M. Tareen, A. Ikram, H. Rahman, A. Wadood, M. Qasim, et al., “Prevalence of HCV among the young male blood donors of Quetta region of Balochistan, Pakistan,” Virology journal, vol. 10, pp. 1-4, 2013.
[3] S. Park, E. Lee, C. Rim, and J. Seong, “Cell-free DNA as a predictive marker after radiation therapy for hepatocellular carcinoma,” International Journal of Radiation Oncology, Biology, Physics, vol. 99, pp. S89-S90, 2017.
[4] L. Cao, B. Yu, D. Kong, Q. Cong, T. Yu, Z. Chen, et al., “Functional expression and characterization of the envelope glycoprotein E1E2 heterodimer of hepatitis C virus,” PLoS pathogens, vol. 15, p. e1007759, 2019.
[5] L. Shekhtman, M. Navasa, N. Sansone, G. Crespo, G. Subramanya, T. L. Chung, et al., “Modeling hepatitis C virus kinetics during liver transplantation reveals the role of the liver in virus clearance,” Elife, vol. 10, p. e65297, 2021.
[6] N. I. o. Health, “Proceedings of the National Institutes of Health Consensus Development Conference Statement-Management of Hepatitis C, 10-12 June 2002,” Hepatology, vol. 36, pp. S3-S20, 2002.
[7] C. S. García-Romero, C. Guzman, A. Cervantes, and M. Cerbón, “Liver disease in pregnancy: Medical aspects and their implications for mother and child,” Annals of hepatology, vol. 18, pp. 553-562, 2019.
[8] S. Niknamian, “MicroRNAs biomarkers profiling in diagnosis and therapeutic management of hepatitis B virus infection,” Aditum Journal of Clinical and Biomedical Research, vol. 2, 2021.
[9] M. A. Khattab, Y. Zakaria, E. Sadek, A. E. Fatah, S. Aliaa, M. Fouad, et al., “Detection of hepatitis C virus (HCV) RNA in the peripheral blood mononuclear cells of HCV-infected patients following sustained virologic response,” Clinical and Experimental Medicine, pp. 1-10, 2022.
[10] F. Bohne, M. Martínez-Llordella, J.-J. Lozano, R. Miquel, C. Benítez, M.-C. Londoño, et al., “Intra-graft expression of genes involved in iron homeostasis predicts the development of operational tolerance in human liver transplantation,” The Journal of clinical investigation, vol. 122, pp. 368-382, 2012.
[11] G. M. Lauer and B. D. Walker, “Hepatitis C virus infection,” New England journal of medicine, vol. 345, pp. 41-52, 2001.
[12] I. Abbadi, M. Lkhider, B. Kitab, K. Jabboua, I. Zaidane, A. Haddaji, et al., “Non-primate hepacivirus transmission and prevalence: Novel findings of virus circulation in horses and dogs in Morocco,” Infection, Genetics and Evolution, vol. 93, p. 104975, 2021.
[13] D. Moskophidis, F. Lechner, H. Pircher, and R. M. Zinkernagel, “Virus persistence in acutely infected immunocompetent mice by exhaustion of antiviral cytotoxic effector T cells,” Nature, vol. 362, pp. 758-761, 1993.
[14] A. M. Grabowska, F. Lechner, P. Klenerman, P. J. Tighe, S. Ryder, J. K. Ball, et al., “Direct ex vivo comparison of the breadth and specificity of the T cells in the liver and peripheral blood of patients with chronic HCV infection,” European journal of immunology, vol. 31, pp. 2388-2394, 2001.
[15] P. Simmonds, J. Bukh, C. Combet, G. Deléage, N. Enomoto, S. Feinstone, et al., “Consensus proposals for a unified system of nomenclature of hepatitis C virus genotypes,” Hepatology, vol. 42, pp. 962-973, 2005.
[16] C. G. Yergin, “Immunoregulation By HCV-generated Myeloid-Derived Suppressor Cells,” University of Virginia, 2016.
[17] T. Von Hahn, J. C. Yoon, H. Alter, C. M. Rice, B. Rehermann, P. Balfe, et al., “Hepatitis C virus continuously escapes from neutralizing antibody and T-cell responses during chronic infection in vivo,” Gastroenterology, vol. 132, pp. 667-678, 2007.
[18] I. Castillo, M. Pardo, J. Bartolomé, N. Ortiz-Movilla, E. Rodríguez-Iñigo, S. d. Lucas, et al., “Occult hepatitis C virus infection in patients in whom the etiology of persistently abnormal results of liver-function tests is unknown,” The Journal of infectious diseases, vol. 189, pp. 7-14, 2004.
[19] J. T. Stapleton, W. N. Schmidt, and L. Katz, “Seronegative hepatitis C virus infection, not just RNA detection,” vol. 190, ed: The University Chicago Press, 2004, pp. 651-652.
[20] N. L. Benali-Furet, M. Chami, L. Houel, F. De Giorgi, F. Vernejoul, D. Lagorce, et al., “Hepatitis C virus core triggers apoptosis in liver cells by inducing ER stress and ER calcium depletion,” Oncogene, vol. 24, pp. 4921-4933, 2005.
[21] M. Pardo, J. López‐Alcorocho, E. Rodríguez‐Iñigo, I. Castillo, and V. Carreno, “Comparative study between occult hepatitis C virus infection and chronic hepatitis C,” Journal of viral hepatitis, vol. 14, pp. 36-40, 2007.
[22] C. E. DeSantis, C. C. Lin, A. B. Mariotto, R. L. Siegel, K. D. Stein, J. L. Kramer, et al., “Cancer treatment and survivorship statistics, 2014,” CA: a cancer journal for clinicians, vol. 64, pp. 252-271, 2014.
[23] F. Kanwal, H. B. El-Serag, and D. Ross, “Surveillance for hepatocellular carcinoma: can we focus on the mission?,” Clinical Gastroenterology and Hepatology, vol. 13, pp. 805-807, 2015.
[24] R. J. Wong, R. Cheung, and A. Ahmed, “Nonalcoholic steatohepatitis is the most rapidly growing indication for liver transplantation in patients with hepatocellular carcinoma in the US,” Hepatology, vol. 59, pp. 2188-2195, 2014.
[25] I. M. Jacobson, G. L. Davis, H. El–Serag, F. Negro, and C. Trépo, “Prevalence and challenges of liver diseases in patients with chronic hepatitis C virus infection,” Clinical Gastroenterology and Hepatology, vol. 8, pp. 924-933, 2010.
[26] J. W. Galbraith, J. P. Donnelly, R. A. Franco, E. T. Overton, J. B. Rodgers, and H. E. Wang, “National estimates of healthcare utilization by individuals with hepatitis C virus infection in the United States,” Clinical infectious diseases, vol. 59, pp. 755-764, 2014.
[27] V. Yakovchenko, R. E. Bolton, M.-L. Drainoni, and A. L. Gifford, “Primary care provider perceptions and experiences of implementing hepatitis C virus birth cohort testing: a qualitative formative evaluation,” BMC health services research, vol. 19, pp. 1-8, 2019.
[28] R. Nawaz, S. Zahid, M. Idrees, S. Rafique, M. Shahid, A. Ahad, et al., “HCV-induced regulatory alterations of IL-1β, IL-6, TNF-α, and IFN-ϒ operative, leading liver en-route to non-alcoholic steatohepatitis,” Inflammation Research, vol. 66, pp. 477-486, 2017.
[29] E. Caparrós and R. Francés, “The interleukin-20 cytokine family in liver disease,” Frontiers in Immunology, vol. 9, p. 1155, 2018.
[30] S. Radaeva, R. Sun, H. n. Pan, F. Hong, and B. Gao, “Interleukin 22 (IL‐22) plays a protective role in T cell‐mediated murine hepatitis: IL‐22 is a survival factor for hepatocytes via STAT3 activation,” Hepatology, vol. 39, pp. 1332-1342, 2004.
[31] L. Yang, Y. Zhang, L. Wang, F. Fan, L. Zhu, Z. Li, et al., “Amelioration of high fat diet induced liver lipogenesis and hepatic steatosis by interleukin-22,” Journal of hepatology, vol. 53, pp. 339-347, 2010.
[32] K. Wolk, H. S. Haugen, W. Xu, E. Witte, K. Waggie, M. Anderson, et al., “IL-22 and IL-20 are key mediators of the epidermal alterations in psoriasis while IL-17 and IFN-γ are not,” Journal of molecular medicine, vol. 87, pp. 523-536, 2009.
[33] S. Meller, J. Di Domizio, K. S. Voo, H. C. Friedrich, G. Chamilos, D. Ganguly, et al., “TH 17 cells promote microbial killing and innate immune sensing of DNA via interleukin 26,” Nature immunology, vol. 16, pp. 970-979, 2015.
[34] E. Stephen-Victor, H. Fickenscher, and J. Bayry, “IL-26: an emerging proinflammatory member of the IL-10 cytokine family with multifaceted actions in antiviral, antimicrobial, and autoimmune responses,” PLoS pathogens, vol. 12, p. e1005624, 2016.
[35] K. Ohnuma, R. Hatano, T. M. Aune, H. Otsuka, S. Iwata, N. H. Dang, et al., “Regulation of pulmonary graft-versus-host disease by IL-26+ CD26+ CD4 T lymphocytes,” The Journal of Immunology, vol. 194, pp. 3697-3712, 2015.
[36] M. E. Menezes, S. Bhatia, P. Bhoopathi, S. K. Das, L. Emdad, S. Dasgupta, et al., “MDA-7/IL-24: multifunctional cancer killing cytokine,” Anticancer Genes, pp. 127-153, 2014.
[37] A. Wieckowska, B. G. Papouchado, Z. Li, R. Lopez, N. N. Zein, and A. E. Feldstein, “Increased hepatic and circulating interleukin-6 levels in human nonalcoholic steatohepatitis,” Official journal of the American College of Gastroenterology| ACG, vol. 103, pp. 1372-1379, 2008.
[38] H. Tilg and A. M. Diehl, “Cytokines in alcoholic and nonalcoholic steatohepatitis,” New England Journal of Medicine, vol. 343, pp. 1467-1476, 2000.
[39] R. C. Lee, R. L. Feinbaum, and V. Ambros, “The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14,” cell, vol. 75, pp. 843-854, 1993.
[40] V. Ambros, “The functions of animal microRNAs,” Nature, vol. 431, pp. 350-355, 2004.
[41] S. Cermelli, A. Ruggieri, J. A. Marrero, G. N. Ioannou, and L. Beretta, “Circulating microRNAs in patients with chronic hepatitis C and non-alcoholic fatty liver disease,” PloS one, vol. 6, p. e23937, 2011.
[42] H. Yamada, K. Suzuki, N. Ichino, Y. Ando, A. Sawada, K. Osakabe, et al., “Associations between circulating microRNAs (miR-21, miR-34a, miR-122 and miR-451) and non-alcoholic fatty liver,” Clinica chimica acta, vol. 424, pp. 99-103, 2013.
[43] S. Bala and G. Szabo, “MicroRNA signature in alcoholic liver disease,” International journal of hepatology, vol. 2012, 2012.
[44] D. Blaya, M. Coll, D. Rodrigo-Torres, M. Vila-Casadesús, J. Altamirano, M. Llopis, et al., “Integrative microRNA profiling in alcoholic hepatitis reveals a role for microRNA-182 in liver injury and inflammation,” Gut, vol. 65, pp. 1535-1545, 2016.
[45] F. Borel, P. Konstantinova, and P. L. Jansen, “Diagnostic and therapeutic potential of miRNA signatures in patients with hepatocellular carcinoma,” Journal of hepatology, vol. 56, pp. 1371-1383, 2012.
[46] Y. Q. Cheng, J. P. Ren, J. Zhao, J. M. Wang, Y. Zhou, G. Y. Li, et al., “Micro RNA‐155 regulates interferon‐γ production in natural killer cells via T im‐3 signalling in chronic hepatitis C virus infection,” Immunology, vol. 145, pp. 485-497, 2015.
[47] O. Cheung, P. Puri, C. Eicken, M. J. Contos, F. Mirshahi, J. W. Maher, et al., “Nonalcoholic steatohepatitis is associated with altered hepatic MicroRNA expression,” Hepatology, vol. 48, pp. 1810-1820, 2008.
[48] S. Bala, M. Marcos, K. Kodys, T. Csak, D. Catalano, P. Mandrekar, et al., “Up-regulation of microRNA-155 in macrophages contributes to increased tumor necrosis factor α (TNFα) production via increased mRNA half-life in alcoholic liver disease,” Journal of Biological Chemistry, vol. 286, pp. 1436-1444, 2011.
[49] M. Lagos-Quintana, R. Rauhut, A. Yalcin, J. Meyer, W. Lendeckel, and T. Tuschl, “Identification of tissue-specific microRNAs from mouse,” Current biology, vol. 12, pp. 735-739, 2002.
[50] M. Girard, E. Jacquemin, A. Munnich, S. Lyonnet, and A. Henrion-Caude, “miR-122, a paradigm for the role of microRNAs in the liver,” Journal of hepatology, vol. 48, pp. 648-656, 2008.
[51] W. Hou, Q. Tian, J. Zheng, and H. L. Bonkovsky, “MicroRNA‐196 represses Bach1 protein and hepatitis C virus gene expression in human hepatoma cells expressing hepatitis C viral proteins,” Hepatology, vol. 51, pp. 1494-1504, 2010.
[52] P. J. Starkey Lewis, J. Dear, V. Platt, K. J. Simpson, D. G. Craig, D. J. Antoine, et al., “Circulating microRNAs as potential markers of human drug‐induced liver injury,” Hepatology, vol. 54, pp. 1767-1776, 2011.
[53] A. J. van der Meer, B. J. Veldt, J. J. Feld, H. Wedemeyer, J.-F. Dufour, F. Lammert, et al., “Association between sustained virological response and all-cause mortality among patients with chronic hepatitis C and advanced hepatic fibrosis,” Jama, vol. 308, pp. 2584-2593, 2012.
[54] B. Clarke, “Molecular virology of hepatitis C virus,” Journal of General Virology, vol. 78, pp. 2397-2410, 1997.
[55] A. Manzin, M. Candela, S. Paolucci, M. Caniglia, A. Gabrielli, and M. Clementi, “Presence of hepatitis C virus (HCV) genomic RNA and viral replicative intermediates in bone marrow and peripheral blood mononuclear cells from HCV-infected patients,” Clinical and diagnostic laboratory immunology, vol. 1, p. 160, 1994.
[56] J.-T. Wang, J.-C. Sheu, J.-T. Lin, T.-H. Wang, and D. S. Chen, “Detection of replicative form of hepatitis C virus RNA in peripheral blood mononuclear cells,” Journal of Infectious Diseases, vol. 166, pp. 1167-1169, 1992.
[57] M. G. Saleh, C. J. Tibbs, J. Koskinas, L. M. Pereira, A. B. Bomford, B. C. Portmann, et al., “Hepatic and extrahepatic hepatitis C virus replication in relation to response to interferon therapy,” Hepatology, vol. 20, pp. 1399-1404, 1994.
[58] T. Chang, K. Young, Y. Yang, H. Lei, and H. Wu, “Hepatitis C virus RNA in peripheral blood mononuclear cells: comparing acute and chronic hepatitis C virus infection,” Hepatology, vol. 23, pp. 977-981, 1996.
[59] Y. K. Shimizu, A. Iwamoto, M. Hijikata, R. H. Purcell, and H. Yoshikura, “Evidence for in vitro replication of hepatitis C virus genome in a human T-cell line,” Proceedings of the National Academy of Sciences, vol. 89, pp. 5477-5481, 1992.
[60] Y. K. Shimizu, H. Igarashi, T. Kiyohara, M. Shapiro, D. C. Wong, R. H. Purcell, et al., “Infection of a chimpanzee with hepatitis C virus grown in cell culture,” Journal of General Virology, vol. 79, pp. 1383-1386, 1998.
[61] C. Feray, D. Samuel, V. Thiers, M. Gigou, F. Pichon, A. Bismuth, et al., “Reinfection of liver graft by hepatitis C virus after liver transplantation,” The Journal of clinical investigation, vol. 89, pp. 1361-1365, 1992.
[62] T. Laskus, M. Radkowski, J. Wilkinson, H. Vargas, and J. Rakela, “The origin of hepatitis C virus reinfecting transplanted livers: serum-derived versus peripheral blood mononuclear cell-derived virus,” The Journal of infectious diseases, vol. 185, pp. 417-421, 2002.
[63] I. Castillo, E. Rodriguez-Inigo, J. Bartolome, S. De Lucas, N. Ortiz-Movilla, J. López-Alcorocho, et al., “Hepatitis C virus replicates in peripheral blood mononuclear cells of patients with occult hepatitis C virus infection,” Gut, vol. 54, pp. 682-685, 2005.
[64] G.-Z. Gong, L.-Y. Lai, Y.-F. Jiang, Y. He, and X.-S. Su, “HCV replication in PBMC and its influence on interferon therapy,” World Journal of Gastroenterology: WJG, vol. 9, p. 291, 2003.
[65] F. A. Di Lello, A. C. A. Culasso, C. Parodi, P. Baré, R. H. Campos, and G. García, “New evidence of replication of hepatitis C virus in short-term peripheral blood mononuclear cell cultures,” Virus research, vol. 191, pp. 1-9, 2014.
[66] P. Baré, “Hepatitis C virus and peripheral blood mononuclear cell reservoirs Patricia Baré,” World journal of hepatology, vol. 1, p. 67, 2009.
[67] A. Alhetheel, A. Albarrag, A. Hakami, Z. Shakoor, K. Alswat, A. Abdo, et al., “In the peripheral blood mononuclear cells (PBMCs) of HCV infected patients the expression of STAT1 and IRF-1 is downregulated while that of caspase-3 upregulated,” Acta virologica, vol. 64, pp. 352-358, 2020.
[68] L. Deng, T. Adachi, K. Kitayama, Y. Bungyoku, S. Kitazawa, S. Ishido, et al., “Hepatitis C virus infection induces apoptosis through a Bax-triggered, mitochondrion-mediated, caspase 3-dependent pathway,” Journal of virology, vol. 82, pp. 10375-10385, 2008.
[69] T. Marcello, A. Grakoui, G. Barba–Spaeth, E. S. Machlin, S. V. Kotenko, M. R. Macdonald, et al., “Interferons α and λ inhibit hepatitis C virus replication with distinct signal transduction and gene regulation kinetics,” Gastroenterology, vol. 131, pp. 1887-1898, 2006.
[70] S. V. Kotenko, G. Gallagher, V. V. Baurin, A. Lewis-Antes, M. Shen, N. K. Shah, et al., “IFN-λs mediate antiviral protection through a distinct class II cytokine receptor complex,” Nature immunology, vol. 4, pp. 69-77, 2003.
[71] S. Cooper, A. L. Erickson, E. J. Adams, J. Kansopon, A. J. Weiner, D. Y. Chien, et al., “Analysis of a successful immune response against hepatitis C virus,” Immunity, vol. 10, pp. 439-449, 1999.
[72] O. Waidmann, V. Bihrer, T. Pleli, H. Farnik, A. Berger, S. Zeuzem, et al., “Serum microRNA‐122 levels in different groups of patients with chronic hepatitis B virus infection,” Journal of viral hepatitis, vol. 19, pp. e58-e65, 2012.
[73] S. J. Weston, R. L. Leistikow, K. R. Reddy, M. Torres, A. M. Wertheimer, D. M. Lewinsohn, et al., “Reconstitution of hepatitis C virus–specific T‐cell–mediated immunity after liver transplantation,” Hepatology, vol. 41, pp. 72-81, 2005.